Single-cell RNA-sequencing is increasingly used to map the cellular heterogeneity of complex tissues. For example, more than 300 studies profiled a total of over 44 million cells across different brain regions, ages, and species. Since different research groups usually specialize in different cell-type compartments, these datasets usually differ in terminology and annotation depth. Our understanding of cellular diversity can be greatly enhanced by integrating these datasets to obtain a comprehensive atlas. In a recent publication, Lieke Michielsen, Marcel Reinders and Ahmed Mahfouz, in collaboration with the group of Fabian Theis (Helmholtz Centrum München), present ‘treeArches’, a framework to automatically build and update such reference atlases with a corresponding cell-type hierarchy. The atlas can be used to transfer cell annotations to new unlabeled datasets, facilitating reproducible analysis.
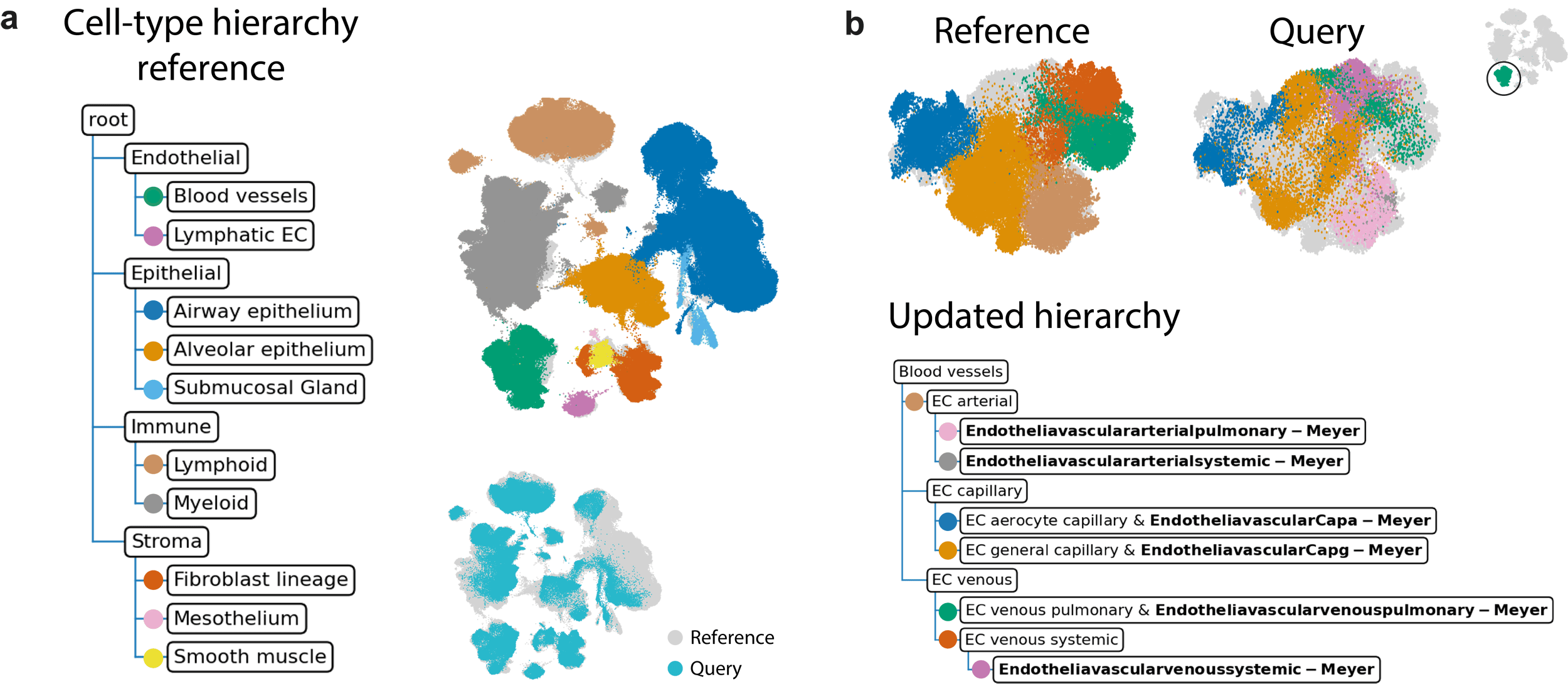
In order to develop such atlas, Michielsen et al. had to take into account two major challenges. First, identifying relationships between cell types across datasets is challenging. How does a cell type from study A match a cell type from study B? These identities are assigned by different researchers and the process is very subjective. Even more problematic is the fact that there is no universal convention on cellular hierarchy. For example, while one study can label cells as neurons, another differentiates between excitatory and inhibitory neurons, which in fact is a sub-compartment of the former. Second, as more single-cell data becomes available, this atlas needs to be updated which renders previous analysis using an older version of the atlas obsolete. Ideally, a new dataset would be projected on the existing atlas, such that the atlas can be updated progressively instead of remade from scratch. To overcome these challenges, treeArches combines two existing methods: scHPL and scArches. scHPL tackles the first challenge of matching the cell types across datasets and automatically builds a cell-type hierarchy. Using scArches, new datasets can be projected on top of the reference atlas such that the atlas and the corresponding hierarchy can be updated. If the cells in the new datasets are unlabeled, the current atlas can also be used to automatically annotate the cells. In the paper, they apply treeArches amongst others to the Human Lung Cell Atlas, a reference atlas that consists of 16 lung datasets with a well-defined cell-type hierarchy. They have chosen this example as a proof-of-concept because of the high quality annotations of this atlas. When projecting new datasets on this atlas, the authors show that they can either increase the resolution of the hierarchy when these datasets are labeled, or can annotate the cells if the dataset is unlabeled. They could also detect diseased cell types in an idiopathic pulmonary fibrosis dataset.

Updated hierarchy when adding annotations of the Meyer et al. (2023) dataset to the reference atlas.
Even though the authors mainly focus on lung datasets in the paper, this tool can be applied to any single-cell RNA-sequencing dataset regardless of the tissue type. They envision that treeArches will enable a data-driven path towards consensus-based cell type annotation of (human) tissues and will significantly speed up the building and annotation of atlases. Within BRAINSCAPES, there is a specific interest in the reward system including the VTA. Researchers from the Basak lab are already testing whether this tool can help them create a reference atlas for this brain region. More specifically, they are comparing the human and mouse VTA datasets to publicly available datasets that are derived from partially overlapping brain regions, with different cellular composition, resolution and annotation, to unify the literature.
The full article can be read here: Single-cell reference mapping to construct and extend cell-type hierarchies | NAR Genomics & Bioinformatics.